Background
While most cells in multicellular organisms carry the same genetic information, in each cell type only a subset of genes is being transcribed. Such differentiation in gene expression depends, for a large part, on the activation and repression of regulatory sequences, including transcriptional enhancers. Transcriptional enhancers can be located tens of kilobases from their target genes, but display characteristic chromatin and DNA features, allowing their identification by genome-wide profiling. Here we show that integration of chromatin characteristics can be applied to predict distal enhancer candidates in Zea mays, thereby providing a basis for a better understanding of gene regulation in this important crop plant.
Result
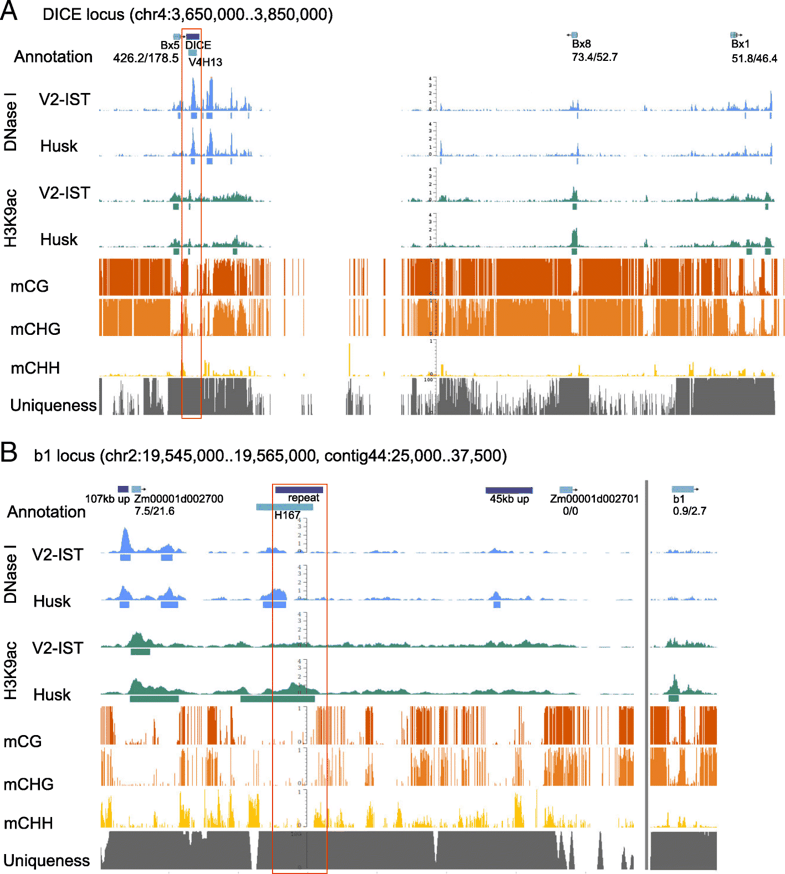
To predict transcriptional enhancers in the crop plant maize (Zea mays L. ssp. mays), we integrated available genome-wide DNA methylation data with newly generated maps for chromatin accessibility and histone 3 lysine 9 acetylation (H3K9ac) enrichment in young seedling and husk tissue. Approximately 1500 intergenic regions, displaying low DNA methylation, high chromatin accessibility and H3K9ac enrichment, were classified as enhancer candidates. Based on their chromatin profiles, candidate sequences can be classified into four subcategories. Tissue-specificity of enhancer candidates is defined based on the tissues in which they are identified and putative target genes are assigned based on tissue-specific expression patterns of flanking genes.
Conclusions
Our method identifies three previously identified distal enhancers in maize, validating the new set of enhancer candidates and enlarging the toolbox for the functional characterization of gene regulation in the highly repetitive maize genome.